Le Infezioni in Medicina, n. 4, 476-487, 2023
doi: 10.53854/liim-3104-6
REVIEWS
Interaction of the viral infectious agents in the development and exacerbation of the multiple sclerosis
Sina Mahdavi1, Mahdi Asghari Ozma2, Arezou Azadi1, Javid Sadeghi1, Hossein Bannazadeh Baghi3, Mahin Ahangar Oskouee3
1Department of Microbiology and Virology, Tabriz University of Medical Sciences, Tabriz, Iran;
2Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz, Iran;
3Infectious and Tropical Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
Article received 8 August 2023; accepted 28 October 2023
Corresponding author
Mahin Ahangar Oskouee
E-mail: ahangar1342@gmail.com
SummaRY
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disorder of the central nervous system (CNS). The underlying cause of MS is still unknown. Multiple risk factors have been suggested that involve a combination of genetic, environmental, and infectious factors that contribute along with a weakened immune system. There is growing evidence supporting the potential role of viral infections in the development of the disease. Viruses like human immunodeficiency virus (HIV), John Cunningham virus (JCV), Varicella-Zoster virus (VZV), human herpesvirus 6 (HHV-6), Epstein-Barr virus (EBV), and human endogenous retroviruses (HERVs) have been proposed in the pathogenesis of MS. Their pathogenetic mechanisms are not well known, but several possibilities have been discussed. The present study highlights the proposed potential molecular and genetic mechanisms underlying this viral interaction and its implications for the development of MS.
Keywords: Epstein-Barr Virus, Human Endogenous Retrovirus, Human Immunodeficiency Virus, John Cunningham Virus, Multiple Sclerosis.
INTRODUCTION
Multiple sclerosis (MS) is a chronic progressive inflammatory disorder that affects the central nervous system’s (CNS), which consists of the brain, optic nerves, and spinal cord [1]. It generally affects people between the ages of 20 and 40 and is the most prevalent neurological illness that causes disability in young adults. According to estimates, 2.8 million individuals worldwide are affected by MS, with a 35.9 per 100,000 population prevalence rate [2, 3]. The immune system wrongly targets myelin, the protective layer that protects nerve fibers, in MS. This breakdown of myelin results in scar tissue known as sclerosis, which can also be referred to as plaques or lesions [4]. Myelin is a multi-layer sheath formed by neuroglial cells around neurons and axons [5]. The myelin sheath allows nerve impulses to move quickly and effectively along nerve cells. When myelin degeneration occurs, these impulses decelerate [6].
The underlying pathology seems to be focal demyelination and neurodegeneration of the CNS [7]. Plaques have been identified in the damaged regions, which are mostly supported by the white matter that encircles the lateral ventricles of the brain and the optic nerves. Most of the time, conventional magnetic resonance imaging (MRI) techniques can find demyelination in the white matter and gray matter of a person with MS [8]. The degree of cortical demyelination is thought to reflect the clinical progression of MS, whereas gray matter demyelination is correlated with a progressive form of the illness [8, 9].
MS affects people differently, with some experiencing only mild symptoms while others may include sensory and motor problems, visual abnormalities, and cognitive impairment. Primary progressive MS (PPMS), secondary progressive MS (SPMS), relapsing-remitting MS (RRMS), and progressive relapsing MS (PRMS) are the four clinical phases of MS [10]. The most frequent form of the condition, RRMS, is characterized by clinical symptoms that develop following periods of partial or complete remission. SPMS is a secondary progressive course of RRMS with sustained deterioration and advancement of symptoms [11]. In PPMS, unlike PRMS, in which the period coincides with the progression of MS symptoms, there is no improvement during the continuous progression of neurological symptoms. Improvement also appears. Several treatments help improve symptoms, but there is no improvement in some people, and the condition may progress to SPMS [12].
A multifactorial hypothesis has been postulated, in which complex combinations of numerous “environmental or infectious” elements may operate as triggers that cause autoimmunity and disease progression, despite the fact that several parts of the etiology and pathophysiology of MS remain unexplained [13, 14]. MS risk factors include ethnicity, particularly human leukocyte antigens (HLA) loci, the most significant of which is HLA-DRB1 *15:01 in the class II region, gender (more prevalent in women), latitude (and consequently sunshine and vitamin D availability), and viral infections [7]. Various immunomodulatory therapies are used to manage symptoms, accelerate recovery after relapses, and partially reduce the number and severity of relapses. None of them, however, has the ability to halt or slow the progression of the disease entirely [15].
Several different viruses have been proposed to be associated with the pathogenesis of MS, including human immunodeficiency virus (HIV), John Cunningham virus (JCV), Varicella-Zoster virus (VZV), human herpesvirus 6 (HHV-6), and Epstein-Barr virus (EBV) [16-19]. Moreover, human endogenous retroviruses (HERVs) are discussed [20]. Similar to a portion of the clinical course of MS, herpesvirus infection is followed by a period of remission followed by deteriorating symptoms. However, unlike PPMS, most herpesvirus reactivations do not result in permanent tissue damage or functional loss [21].
Viral infections are one of the aspects of MS that are not fully understood, but they are suspected to play a role in the disease’s pathogenesis. Despite coming from various virus families, these viruses have the capacity to alter the host’s gene expression, bystander activation, epitope release, survival and immortalization of self-stimulating immune cells, and regulatory changes [22]. These viruses have also the ability to pass the blood-brain barrier (BBB) and lead to persistent infections that last a lifetime and can stimulate or inhibit immune pathways, and this dual role can have significant implications for the progression of MS. This potentially leads to immune response dysregulation following myelin destruction and inflammation [23]. The present study written by a considerable number of related articles from online databases such as PubMed, Scopus, Google Scholar, and Web of Science published by reliable publishers such as Springer, Frontiers, and Elsevier from recent years, which were collected and discussed, provides comprehensive information about viral infection in MS disease progression.
THE RELATIONSHIP BETWEEN MS AND HIV
The relationship between HIV and MS has been a topic of interest in recent years. For instance, it has been proposed that there is a connection between HIV infection, leukoencephalomyelopathy, and MS-like disease in some people [24]. The MS-like disease induced by HIV is neuromyelitis optica (NMO), a severe demyelinating disease different from MS that has been observed in HIV-infected persons and requires accurate diagnosis [25]. Berger et al. presented a series of 7 individuals with “MS-like disease” after HIV infection for the first time in 1989 [26]. This was followed by other independent cohorts of acute “MS-like disease” after HIV infection. These groups suggested a little lag between HIV infection and the onset of MS-like symptoms in affected people [27, 28]. Graber and colleagues identified histologic evidence of “MS-like illness” in the presence of HIV, including foamy macrophages, perivascular infiltration with inflammatory cells, and extensive demyelination with reactive astrocytes, all of which are consistent with MS lesions [27]. Published cases have reported the occurrence of MS-like diseases mainly during acute HIV infection or HIV seroconversion [24].
Several autoimmune pathways have been proposed as potential mechanisms in studies on HIV-infected individuals, including:
a) molecular mimicry processes following HIV-1 action on neurons [29];
b) HIV-induced cytokine release that induces polyclonal activation of cells B [30];
c) the appearance of autoantibodies against myelin [31];
d) the loss of regulatory CD8+ T cells [32]; and
e) the apparent restoration of immunity after Highly Active Antiretroviral Therapy (HAART) induction, HAART combines two or more antiretroviral drugs that work against different proteins involved in the HIV life cycle [30].
Hypergammaglobulinemia and prolonged activation of T and B cells are hallmarks of the immune system dysfunction brought on by HIV. When B cells are stimulated, they produce more immunoglobulin, which results in a surplus of Free Light Chains (FLCs). FLCs may be implicated in neuroautoimmune reactions [33]. This suggests that the FLCs may overlap with HIV-associated neurocognitive disorder and MS [34]. Acute HIV infection results in a cytokine storm, a profound release of pro-inflammatory cytokines that contributes to the initial activation of the immune system [35]. Since HLA-DR+, CD38+, CD4+, and CD8+ (activated) T cells have a greater capacity to migrate and affect tissues, such as the brain, expanding their pool can directly raise the risk of developing autoimmune pathology [36].
Epidemiological studies, however, have shown a negative correlation between HIV infection and MS, with HIV-positive individuals having a lower probability of contracting the disease [25, 37, 38]. In a comprehensive study conducted by Maruszak et al. in Denmark, the incidence rate ratio (IRR) of MS among individuals diagnosed with HIV was 0.3 (95% confidence interval [CI] 0.04 to 2.20). However, the statistical analysis did not yield a significant association between MS and HIV in this study [38]. Another research by Gold et al. confirmed a substantial inverse relationship between HIV and MS, with a rate ratio (RR) of 0.38 (95% CI 0.15 to 0.79) of MS incidence in HIV patients compared to controls [39]. In the context of MS, the exact mechanisms by which HIV stimulates and inhibits immune system pathways are not fully understood. However, the persistent immune activation caused by HIV may contribute to the chronic inflammation observed in MS, which is a key factor in the disease’s progression. On the other hand, the inhibition of certain immune system pathways by HIV may contribute to the immune system’s failure to adequately combat MS lesions, leading to disease progression. Furthermore, clinical studies have shown that HAART-treated MS patients with HIV infection had less severe clinical outcomes even when they were not receiving MS medication [25, 40]. It is also possible that HIV itself protects against MS in some way by suppressing HIV expression rather than HAART HIV-induced immunity is expected to lower the incidence of MS because the disease is brought on by autoreactive T cells infecting and demyelinating the CNS. The preventive effects of HAART on the development and course of MS may also contribute to the inverse relationship between HIV and MS [24].
The phase II clinical trial (INSPIRE), which evaluated the effect of the antiretroviral therapy raltegravir on individuals with RRMS, was unsuccessful in demonstrating any advantageous benefits at the group level [41]. Recent clinical research, suggests the possibility that MS treatments may benefit from HIV therapies [24].
THE RELATIONSHIP BETWEEN MS AND JCV
JCV is a nonenveloped, neurotropic, double-stranded DNA virus that is ubiquitous and exclusively human, infecting 80% of the population worldwide. It belongs to the Polyomaviridae family [42]. Primary JCV infection occurs in childhood and remains latent for years in kidney cells, bone marrow, and lymphoid tissue [43]. JCV binds to cellular histones and forms minichromosomes in infected cells. In individuals with a functioning immune system, JCV infection does not result in overt disease, resulting in asymptomatic infection [44]. However, JCV may reactivate in patients undergoing immunosuppressive or immunomodulatory treatments, putting them at a higher risk of developing progressive multifocal leukoencephalopathy (PML), which can result in PML, A rare lytic infection of neuroglial cells, especially oligodendrocytes and astrocytes, causes demyelination [43].
JCV enters host cells by attaching to particular receptors, including integrin molecules, the serotonin receptors, very late antigen-4 (VLA-4), followed by increased proliferation of B cells that cause JCV to enter the brain. However, some patients are susceptible to PML, while others are resistant; mechanisms such as differential reactivity of the patient’s immune system and variation in the virus-host interaction have been suggested [45]. In light of this, it appears necessary to positively evaluate anti-JCV antibody serum as a significant risk factor for PML development before deciding the treatment course for MS patients, despite the fact that natalizumab has a very effective treatment response in RRMS patients [46]. Natalizumab, a monoclonal antibody targeting α4β1 and α4β7 integrins on the surface of lymphocytes, thus inhibits lymphocyte migration [47]. In MS patients, monoclonal antibody immunologic therapy reactivates JCV, a condition for which there is no known therapeutic treatment. It may then generate PML in those who are susceptible, which can exacerbate the disease or result in patient death [48].
THE RELATIONSHIP BETWEEN MS AND VZV
VZV is a neurotropic virus of the herpesvirus family, exclusively human, with almost global distribution. A highly contagious infection in children, the initial infection of which causes varicella (chicken pox), then remains hidden in the cells of the dorsal ganglion of the sensory nerve [49]. Years later, the reactivation of VZV in the form of herpes zoster is more common in people with reduced cellular immunity, especially in the elderly and immunocompromised [50]. Herpes zoster is associated with a variety of neurological syndromes, including postherpetic neuralgia and myelopathy [51].
Epidemiological studies have revealed a significant prevalence of varicella and zoster in MS patients’ clinical antecedents. When MS patients experience an ephemeral relapse, laboratory tests have revealed significant amounts of VZV DNA in their leukocytes and cerebrospinal fluid. However, the virus vanishes during remission [52]. According to research by Sotelo et al., cerebrospinal fluid (CSF) samples from RRMS patients who were in the first week of a clinical relapse included a significant amount of VZV DNA. Patients with MS who were in remission had significantly less VZV DNA in their CSF samples than those who were in relapse. The authors came to the conclusion that VZV has a direct role in the etiology of MS relapse since it is present in the blood and CSF of MS patients who are experiencing relapse and decreases during disease remission [53]. In several studies, it has been shown that most patients with MS in the exacerbation stage of the disease, especially the RRMS stage, have a positive MRZ reaction (MRZR), an intraspinal humoral response to measles (M), rubella (R) and varicella zoster (Z), The MRZR is considered a highly specific marker for MS, meaning it can help distinguish MS from other diseases that may have similar symptoms. For instance, it can help distinguish MS from rheumatologic disorders with central nervous involvement (RDwCNS) [54].
Although the role of VZV infection in MS is still unclear, two models for its infection in patients are proposed [55]. The primary role of VZV glycoprotein E (gE), a major VZV glycoprotein, is to attach to mannose-6-phosphate receptors that aid virus entry. A comparison of the amino acid sequences of HNRNPA1 and VZV proteins revealed a 62% amino acid sequence match between VZV gE and the PrLD/M9 epitope area (TNPO1 binding domain) of mutant HNRNPA1. HNRNPA1 generates a heterogeneous nuclear ribonucleoprotein (hnRNP) that is involved in the processing and transport of mRNA and pre-mRNA, which are found in protein-bound RNAs [56]. Mutant HNRNPA1 mimics VZV gE as an antigen, resulting in autoantibody synthesis. After the accumulation of mutant HnRNPA1, it is transported to the cytoplasm by MHC class I and then activates T lymphocytes. In genetically predisposed people with mutations in the domain PrLD/M9 HnRNPA1, antibodies and immune cells against the gE epitopes of VZV are still present as a result of the memory immunological response, leading to neurodegeneration and the development of RRMS [55, 56].
In September 2010, the US Food and Drug Administration authorized fingolimod (FTY) as the first therapy regimen for RRMS. Although these MS patients are treated with FTY, they have shown an increased incidence of VZV infections. By reducing sphingosine 1-phosphate receptors, fingolimod reduces the migration of lymphocytes and then leads to the progression of VZV disease [57, 58]. These findings, along with VZV’s unique characteristics, such as neurotropism and lengthy periods of latency followed by viral reactivation, support the theory that VZV plays a role in the genesis of MS. But contradictory findings from studies that confirmed the presence of viral gene products in brain tissue point to the need for more investigation into the potential role of VZV in the genesis of MS [52].
THE RELATIONSHIP BETWEEN MS AND HHV-6
HHV-6 consists of two subtypes (HHV-6A and HHV-6B), is acquired in early life, and is widespread across the human population. It is acquired during infancy. The primary infection is usually self-limiting, but once the primary infection has resolved, HHV-6 remains latent in peripheral blood mononuclear cells (PBMCs) and especially HHV-6A in oligodendrocytes [59, 60]. In the early 1990s, the relationship between HHV-6 and MS was proven by higher antibody titers against HHV-6 in MS patients and the finding of elevated HHV-6 DNA proteins in MS plaques in histological studies [60]. HHV-6 has a tropism towards TCD 4+ lymphocytes and enters the CNS due to the weakening of the blood-brain barrier due to inflammatory damage. Therefore, in patients with MS, the entry of HHV-6 into the CNS is increased through the following mechanisms, which can be in demyelination and lead to aggravation of the disease [61]. Identification of HHV-6 DNA in various types of mucous cells and nasal passages showed that this virus could also enter the brain through the olfactory pathway [62].
In the study of Tejada-Simon et al., following the observation that the HHV-6 U24 protein has a 7-amino acid sequence homology with myelin basic protein, which is one of the main components of the myelin sheath, they suggested that it could cause a molecular mimicry mechanism followed by cross-reaction. Interactions between virus and host proteins, including the direct targeting of oligodendrocytes (cells responsible for making myelin sheath) during autoimmune reactions [63]. Reactivation of HHV-6 in the CNS can cause the release of pro-inflammatory cytokines, including TNF-α, which leads to immune-mediated demyelination in patients with MS. Oligodendrocyte progenitor cells expressing viral transcripts, including the U94 gene, had a delay during the HHV-6 incubation period and were not able to migrate, which led to disruption of the myelin repair process. Demyelination caused by HHV-6 may lead to MS exacerbation only in combination with other genetic and environmental risk factors [64] (Figure 1).
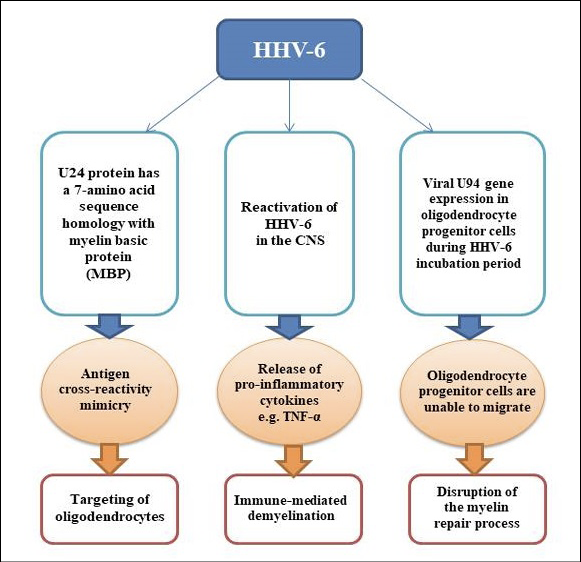
Figure 1 - HHV-6 related mechanisms in MS.
THE RELATIONSHIP BETWEEN MS AND EBV
The Epstein-Barr Virus (EBV) is a member of the herpesviridae family and is transmitted through saliva. It primarily infects B cells and epithelial cells, and can produce a latent infection in B lymphocytes [65]. In an epidemiological investigation of the relationship between MS risk and EBV, the risk of having MS was found to rise 32-fold in EBV-positive individuals [66]. In other studies, both MS patients and healthy individuals have elevated EBV anti-nuclear antibodies, indicating an increased risk of MS. Additionally, demyelinated lesions isolated from MS patients contain EBV nuclear antigens (EBNAs) [67]. The EBNA1 domain contains a pentapeptide fragment similar to αB-crystallin, a heat shock peptide. Peripheral B cells get activated in response to αB-crystallin infection, leading to myelin-directed autoimmunity mediated by proinflammatory T cells [68].
Alleles are variations of a gene that can influence the characteristics of an organism. In this way, they can play a role in the context of human health. The HLA-DR15 is the most potent genetic risk factor in the development of MS pathogenicity, which can act as an auxiliary receptor for EBV entry into B cells [69]. A 7-fold increase in the risk of MS has been observed in EBV infection with HLA-DR15 synergy [70]. The HLA-DRB*07 genotypes in MS patients are associated with higher EBV viral loads, while the HLA-A*02 genotype is associated with lower viral loads. While these findings suggest a potential role for HLA-DRB*07 and HLA-A*02 in the pathogenesis of MS, they do not directly indicate an association with EBV [67]. EBNA2, which regulates viral transcription, may enhance transcription from MS risk loci [71].
In the pathogenesis of MS, the existence and participation of EBV-specific T cells have been reported, including specific TCD4 directed against the EBV DNA-polymerase protein, which subsequently activates myelin basic protein (MBP)-specific CD4+ T cells [67]. Another specific T cell is TH1, specific against EBNA1; Its increase has been found in MS patients [72]. EBV-infected B cells that are also T-bet+ can activate Th17.1 cells, which can infiltrate the CNS in MS patients. The expression of chemokine receptors (CXCR3, CCR6) and adhesion molecules (VLA-4) have been determined. Within the CNS, Th17.1 cells produce IFN-γ, and GM-CSF, together with T-bet+ memory B cells, come into contact with follicle-like structures and then cause inflammation, clonal expansion, and cytotoxic potential of demyelination. T-bet+ memory B cells differentiate into plasmablasts and secrete a large number of autoantibodies [73]. EBV-infected cells can act as APCs and lead to the stimulation of EBV cross-reactive CD4+ T cells, which cause inflammatory damage [74]. Due to the chronic infection of B cells infected with EBV, the continuous presentation of antigens leads to disruption and reduction of cell function, known as T cell exhaustion [69]. EBV infection, along with a variety of specific genetic risk alleles, causes inflammatory cascades in the CNS by infected B cells and anti-EBV specific T cells, especially Th17 [75] (Table 1).
Table 1 - The association between EBV and MS.

Based on the success of studies targeting CD20, which reduces MS relapse and lesion formation, the essential role of B cells in the pathogenesis of MS has been established [76]. Among other things, the treatment regimen against EBV has been studied on MS patients: Anti-CD52 mAb and cladribine acted as B cell suppressors [69]. Teriflunomide reduces lymphoid proliferation caused by EBV [77]. The non-cyclic nucleoside analog tenofovir alafenamide (TAF), after inhibiting the activity of EBV DNA polymerase 224 and 225, inhibits EBV replication. These treatment regimens showed clinical benefits and reduced the severity of MS disease, but treatments such as atacicept that target naive and plasma B cells it has caused the exacerbation of MS disease [67]. Understanding the relationship between EBV and MS has implications for the development of targeted therapies, such as antivirals and vaccines, to treat and potentially prevent MS [78]. However, it is essential to note that most people with EBV do not develop MS, and the exact role of EBV in the development of the disease remains to be elucidated [78].
THE RELATIONSHIP BETWEEN MS AND HERV
In 1989, Peron et al. identified a novel retroviral-like element associated with reverse transcriptase (RT) activity in leptomeningeal cells of MS patients and named it multiple sclerosis-associated retroviruses (MSRV) [79]. Laufer et al. demonstrated that all of the MSRV envelope (env) sequences were derived from HERV-W env Xq22.3 [80]. Approximately 8–9% of the human genome is made up of HERVs [81, 82]. However, in normal tissues and cultured cells, between 7 and 30% of all HERV sequences in the genome may be transcriptionally active [83]. Epigenetic and Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like (APOBEC) mechanisms contribute to the regulation of HERV expression in the genome. Epigenetics is a modify the activation of certain genes, but not the genetic code sequence of DNA. The microstructure of DNA itself or the associated chromatin proteins may be modified, causing activation or silencing. APOBEC enzymes play key roles in innate immunity with function cytidine deaminases, helping to prevent viral infections and control retrotransposons [84]. External events, especially viral infections such as EBV, herpes simplex virus type1 (HSV-1), and influenza virus, can act as activators of HERV expression [85, 86]. In a study, it has been shown that the binding of EBV surface glycoprotein, called gp350, can activate the expression of MSRV/HERV-W in peripheral blood cells and astrocytes [87].
Despite attempts to suppress HERVs expression, they can still be expressed in the human CNS due to a combination of epigenetic mechanisms and environmental factors. MRSV/HERV-W, HERV-Fc1, and HERV-K-18 are mostly linked to an increased risk of developing MS [87]. The first evidence of MSRV/HERV-W expression in PBMCs from MS patients was reported by Brudek and colleagues [87]. Additionally, several investigations have shown that HERV-W/MRSV expression in B lymphocytes, monocytes, and natural killer (NK) cells is present in RRMS patients [88-90]. MSRVEnv can activate TLR4 at the brain surface, specifically in oligodendroglial progenitor cells and macrophages, leading to immune cascades, including the production of pro-inflammatory cytokines and inducible nitric oxide synthase (iNOS), followed by the reduction of myelin protein expression [91]. This viral protein has been detected in microglia/macrophages in chronic active MS brain lesions in TLR4-positive oligodendroglial progenitor cells [91]. Superantigen encoded by the HERV-K18 env is transactivated by EBV proteins LMP-2A and CD21 via ITAM (immune receptor tyrosine-based activation motif) infected B cells [92].
HERV-W env has a cell receptor called sodium-dependent neutral amino acid transporter type 1 and type 2 (ASCT-1 and ASCT-2), which is mainly found in human astrocytes [93]. The survival of myelin and neurons depends on L-serine, which is mainly transmitted through the ASCT-1 receptor, which is suppressed in the white matter of MS patients. The binding of HERV-W to this receptor induces the expression of the endoplasmic reticulum stress sensor and inducible nitric oxide synthase (iNOS) sensor, leading to oxidative stress [92]. Revealed higher levels of syncytin-1 expression, an envelope glycoprotein of HERV-W that has a role in cell fusion, in glial cells of MS lesions in demyelinated regions of MS patients’ brains [94]. An increase in syncytin-1 levels leads to an increase in the inflammatory cytokine IL-1β and also through the induction of old astrocyte-specific inducing substance (OASIS, an endoplasmic reticulum stress sensor) leading to an increase in iNOS and by inducing the release of reactive oxygen species (ROS) redox causes cytotoxicity of oligodendrocytes and demyelination [95] (Figure 2).

Figure 2 - (A) Mechanisms of silencing and expression of HERV genes. (B) Possible pathways of HERVs effect on MS susceptibility and progression. Abbreviation: pol (polymerase), gag (group-specific antigen), env (envelope), LTR (long terminal repeat), APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide), TLR-4 (Toll-like receptor 4), ASCT-1 (Alanine/Serine/Cysteine/Threonine-preferring Transporter 1), IL-1β (interleukin 1 beta), iNOS (Inducible nitric oxide synthase), ROS (reactive oxygen species).
There is a high expression of endogenous retroviruses, especially HERV-W/MSRV, during the course of MS, which shows the relationship between HERV and MS, that this virus can play a role in the progression of MS by creating an inflammatory state in the CNS [96]. The presence of virions, proteins of endogenous retroviruses in blood and cerebrospinal fluid, and MS lesions may be predictive of this disease [87]. The studies showed that targeting HERV-W/MSRV expression in MS patients using natalizumab and interferon (IFN)-β reduced HERV-W/MSRV expression after 3 and 6 months of treatment [88]. Therefore, strategies to control endogenous retrovirus expression may be useful in lowering inflammatory processes and oxidative damage in demyelinated regions of MS patients [87].
IMMUNOMODULATORY THERAPIES AND THEIR POTENTIAL EFFECT ON MS-RELATED VIRAL INFECTIOUS AGENTS
Immunomodulatory therapies are a class of drugs that work by influencing the immune system’s responses. They can suppress the immune system, prevent the immune system from attacking the body’s own tissues, or help the immune system attack cancer cells. In the context of MS, these therapies could potentially be used to reduce inflammation and stop the immune system from attacking the body’s own tissues [97]. In a recent investigation, it was observed that patients with MS were more likely to have a history of viral infections. Furthermore, this study highlighted that viral infections could trigger an inflammatory response, which is a key feature of MS. This suggests that viral infections could potentially be a target for immunomodulatory therapies [98].
In terms of specific immunomodulatory therapies, IFN-β is a commonly used treatment for MS. IFN-β works by blocking the viral proteins that trigger an immune response. This can reduce inflammation and slow the progression of MS [99]. Another immunomodulatory therapy is natalizumab, a monoclonal antibody that works by binding to a protein called aquaporin-4 (AQP4). AQP4 is involved in the immune response and is overexpressed in people with MS. By blocking AQP4, natalizumab can reduce inflammation and slow the progression of MS [100]. The antibody is designed to target two types of integrins: α4β1 and α4β7. In the context of MS, these integrins have been found to play a role in the migration of immune cells into the central nervous system, contributing to the disease’s progression. By blocking these integrins, natalizumab may help to limit the movement of these immune cells and reduce the inflammatory response in the brain [101]. While natalizumab is an effective treatment for MS, it is not without risks. One of the most serious risks associated with natalizumab is the JC virus (JCV), a virus that can cause a rare but potentially serious brain tumor called progressive multifocal leukoencephalopathy (PML). The presence of JCV DNA in the cerebrospinal fluid (CSF) of people with MS has been detected in some patients treated with natalizumab. However, the presence of JCV DNA does not necessarily mean that the patient will develop PML [102].
CONCLUSIONS
MS is a neurodegenerative disease subject to a complex interaction between genetic, environmental, and infectious factors. However, viral infection‘s exact role and mechanisms have not yet been determined. Epidemiological observations showed the presence of antiviral immunity in RRMS patients, and viral components were found in MS lesions. These viral infections may trigger or aggravate the disease through different immune pathways, especially in individuals with suppressed immune system conditions or genetically predisposed people. Viral infections should consider as a risk factor for MS. More studies need to be done to clarify our understanding of possible links between MS and viruses.
Conflict of interest
None.
Funding
This study was supported by Tabriz University of Medical Sciences with grant number 69572 and approved by the local ethic committee.
Acknowledgments
We thank all comments and helps by our colleagues from Infectious and Tropical Diseases Research Center.
REFERENCES
[1] Ford H. Clinical presentation and diagnosis of multiple sclerosis. Clin Med. 2020; 20 (4): 380-383.
[2] Nishanth K, Tariq E, Nzvere FP, et al. Role of smoking in the pathogenesis of multiple sclerosis: a review article. Cureus. 2020; 12 (8): e9564.
[3] Walton C, King R, Rechtman L, et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS. Mult Scler J. 2020; 26 (14): 1816-1821.
[4] Shamaa A, Abdallah AN, Bahr MM, El-Tookhy OS. A review: Multiple Sclerosis Treatment: Current Strategies and Future Hopes. Alex J Vet Sci. 2020; 66 (2): 30-39.
[5] Montani L. Lipids in regulating oligodendrocyte structure and function. Semin Cell Dev Biol. 2021; 112: 114-122.
[6] Chen JF, Wang F, Huang NX, Xiao L, Mei F. Oligodendrocytes and myelin: Active players in neurodegenerative brains? Dev Neurobiol. 2022; 82 (2): 160-174.
[7] Tarlinton RE, Khaibullin T, Granatov E, Martynova E, Rizvanov A, Khaiboullina S. The interaction between viral and environmental risk factors in the pathogenesis of multiple sclerosis. Int J Mol Sci. 2019; 20 (2): 303-319.
[8] van Wageningen TA, Gerrits E, Brouwer N, et al. Distinct gene expression in demyelinated white and grey matter areas of patients with multiple sclerosis. Brain Commun. 2022; 4 (2): fcac005.
[9] Fartaria MJ, Bonnier, G, Roche A, et al. Automated detection of white matter and cortical lesions in early stages of multiple sclerosis. J Magn Reson Imaging. 2016; 43 (6): 1445-1454.
[10] Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. Neurology. 1996; 46 (4): 907-911.
[11] Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008; 31: 247-269.
[12] Lublin FD, Reingold SC, Cohen JA, ET AL. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014; 83 (3): 278-286.
[13] Donati D. Viral infections and multiple sclerosis. Drug Discov Today Dis Models. 2020; 32: 27-33.
[14] Sheykhsaran E, Abbasi A, Baghi HB, et al. Staphylococcus aureus: A bacterial candidate for multiple sclerosis incidence and progression. Rev Med Microbiol. 2022; 33 (4): 212-220.
[15] Madsen C. The innovative development in interferon beta treatments of relapsing-remitting multiple sclerosis. Brain Behav. 2017; 7 (6): e00696.
[16] Makhani N, Banwell B, Tellier R, et al. Viral exposures and MS outcome in a prospective cohort of children with acquired demyelination. Mult Scler J. 2016; 22 (3): 385-388.
[17] Paz SPC, Branco L, de Camargo Pereira MA, Spessotto C, Fragoso YD. Systematic review of the published data on the worldwide prevalence of John Cunningham virus in patients with multiple sclerosis and neuromyelitis optica. Epidemiol Health. 2018; 40: e2018001.
[18] Sotelo J, Ordoñez G, Pineda B, Flores J. The participation of varicella zoster virus in relapses of multiple sclerosis. Clin Neurol Neurosurg. 2014; 119: 44-48.
[19] Bert A, Kap YS, Morandi E, Laman JD, Gran B. EBV infection and multiple sclerosis: lessons from a marmoset model. Trends Mol Med. 2016; 22 (12): 1012-1024.
[20] Morandi E, Tanasescu R, Tarlinton RE, Constantinescu CS, Zhang W, Tench C, Gran B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PloS one. 2017; 12 (2): e0172415.
[21] Meyding-Lamadé U, Strank C. Herpesvirus infections of the central nervous system in immunocompromised patients. Ther Adv Neurol Disord. 2012; 5 (5): 279-296.
[22] Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe. 2013; 13 (4): 379-393.
[23] Niedobitek G, Meru N, Delecluse HJ. Epstein-Barr virus infection and human malignancies. Int J Exp Pathol. 2001; 82 (3): 149-170.
[24] Stefanou MI, Krumbholz M, Ziemann U, Kowarik MC. Human immunodeficiency virus and multiple sclerosis: a review of the literature. Neurol Res Pract. 2019; 1: 1-7.
[25] Delgado SR, Maldonado J, Rammohan KWJJ. CNS demyelinating disorder with mixed features of neuromyelitis optica and multiple sclerosis in HIV-1 infection. Case report and literature review. J Neurovirol. 2014; 20 (5): 531-537.
[26] Berger J, Sheremata W, Resnick L, Atherton S, Fletcher MA, Norenberg MJN. Multiple sclerosis-like illness occurring with human immunodeficiency virus infection. Neurol J. 1989; 39 (3): 324-324.
[27] Graber P, Rosenmund A, Probst A, Zimmerli WJA. Multiple sclerosis-like illness in early HIV infection. AIDS J. 2000; 14 (15): 2411-2413.
[28] Coban A, Akman-Demir G, Ozsut H, Eraksoy MJTN. Multiple Sclerosis–Like clinical and magnetic resonance imaging findings in Human Immunodeficiency Virus Positive-Case. Neurologist J. 2007; 13 (3): 154-157.
[29] Dobson R, Giovannoni G. Multiple sclerosis-a review. Eur J Neurol. 2019; 26: 27-40.
[30] Virot E, Duclos A, Adelaide L, et al. Autoimmune diseases and HIV infection: a cross-sectional study. Medicine (Baltimore). 2017; 96 (4): e5769.
[31] Baranova SV, Dmitrenok PS, Buneva VN, Sedykh SE, Nevinsky GA. HIV-infected patients: Cross site-specific hydrolysis of H3 and H4 histones and myelin basic protein with antibodies against these three proteins. Molecules. 2021; 26 (2): 316.
[32] Tedeschi V, Paldino G, Kunkl M, et al. CD8+ T cell senescence: lights and shadows in viral infections, autoimmune disorders and cancer. Int J Mol Sci. 2022; 23 (6): 3374.
[33] Gudowska-Sawczuk M, Mroczko BJ. Free light chains as a novel diagnostic biomarker of immune system abnormalities in multiple sclerosis and HIV infection. Biomed Res Int. 2019; 2019.
[34] Borjabad A, Volsky DJ. Common transcriptional signatures in brain tissue from patients with HIV-associated neurocognitive disorders, Alzheimer’s disease, and Multiple Sclerosis. J Neuroimmune Pharmacol. 2012; 7 (4): 914-926.
[35] Malekzadeh A, de Geer-Peeters V, De Groot V, Elisabeth Teunissen C, Beckerman H. Fatigue in patients with multiple sclerosis: is it related to pro-and anti-inflammatory cytokines? Dis Markers. 2015; 2015.
[36] Roe C. HIV immunodynamics and multiple sclerosis. J Neurovirol. 2016; 22 (2): 254-255.
[37] Anand P, Saylor D. Multiple sclerosis and HIV: a case of multiple sclerosis-immune reconstitution inflammatory syndrome associated with antiretroviral therapy initiation. Int J STD AIDS. 2018; 29 (9): 929-932.
[38] Maruszak H, Brew B, Giovannoni G, Gold J. Could antiretroviral drugs be effective in multiple sclerosis? A case report. Eur J Neurol. 2011; 18 (9): e110-e111.
[39] Gold J, Goldacre, R, Maruszak H, Giovannoni G, Yeates D, Goldacre M. HIV and lower risk of multiple sclerosis: beginning to unravel a mystery using a record-linked database study. J Neurol Neurosurg Psychiatry. 2015; 86 (1): 9-12.
[40] Chalkley J, Berger JR. Multiple sclerosis remission following antiretroviral therapy in an HIV-infected man. J Neurovirol. 2014; 20 (6): 640-643.
[41] Gold J, Marta M, Meier UC, et al. A phase II baseline versus treatment study to determine the efficacy of raltegravir (Isentress) in preventing progression of relapsing remitting multiple sclerosis as determined by gadolinium-enhanced MRI: The INSPIRE study. Mult Scler Relat Disord. 2018; 24: 123-128.
[42] Kutsuna T, Zheng H, Abdel-Aziz HO, et al. High JC virus load in tongue carcinomas may be a risk factor for tongue tumorigenesis. Virchows Arch. 2008; 452 (4): 405-410.
[43] Pietropaolo V, Prezioso C, Bagnato F, Antonelli G. John Cunningham virus: an overview on biology and disease of the etiological agent of the progressive multifocal leukoencephalopathy. New Microbiol. 2018; 41 (3): 179-186.
[44] Prado, JCM, Monezi TA, Amorim AT, Lino, V, Paladino A, Boccardo E. Human polyomaviruses and cancer: an overview. Clinics (Sao Paulo). 2018; 73 (1).
[45] Achiron A, Miron G, Zilkha-Falb R, et al. Host cell virus entry mechanisms enhance anti-JCV-antibody switch in natalizumab-treated multiple sclerosis patients. J Neurovirol. 2016; 22 (6): 736-746.
[46] Bartsch T, Rempe T, Leypoldt F , et al. The spectrum of progressive multifocal leukoencephalopathy: a practical approach. Eur J Neurol. 2019; 26 (4): 566-e541.
[47] Gubatan J, Keyashian K, Rubin SJ, Wang J, Buckman CA, Sinha S. Anti-integrins for the treatment of inflammatory bowel disease: current evidence and perspectives. Clin Exp Gastroenterol. 2021; 14: 333-342.
[48] Assetta B, Atwood WJ The biology of JC polyomavirus. Biol Chem. 2017; 398 (8): 839-855.
[49] Kennedy PG, Mogensen TH, Cohrs RJ. Recent issues in varicella-zoster virus latency. Viruses. 2021; 13 (10): 2018.
[50] Kennedy PG, Gershon AA. Clinical features of varicella-zoster virus infection. Viruses. 2018; 10 (11): 609.
[51] Nagel MA, Niemeyer CS, Bubak AN. Central nervous system infections produced by varicella zoster virus. Curr Opin Infect Dis. 2020; 33 (3): 273-278.
[52] Sotelo J, Corona T. Varicella zoster virus and relapsing remitting multiple sclerosis Mult Scler Int. 2011; 2011.
[53] Sotelo J, Martínez-Palomo A, Ordoñez G, Pineda B. Varicella-zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Ann Neurol. 2008; 63 (3): 303-311.
[54] Arneth B, Kraus J. Laboratory biomarkers of multiple sclerosis (MS). Clin Biochem. 2022; 99: 1-8.
[55] Rice EM, Thakolwiboon S, Avila M. Disorders R. Geographic heterogeneity in the association of varicella-zoster virus seropositivity and multiple sclerosis: A systematic review and meta-analysis. Mult Scler Relat Disord. 2021; 53: 103024.
[56] Kattimani Y, Veerappa AM. Complex interaction between mutant HNRNPA1 and gE of varicella zoster virus in pathogenesis of multiple sclerosis. Autoimmunity. 2018; 51 (4): 147-151.
[57] Kappos, L, Radue E-W, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010; 362 (5): 387-401.
[58] Manouchehrinia A, Tanasescu R, Kareem H, et al. Prevalence of a history of prior varicella/herpes zoster infection in multiple sclerosis. J Neurovirol. 2017; 23 (6): 839-844.
[59] Cruz-Muñoz ME, Fuentes-Pananá EM. Beta and gamma human herpesviruses: Agonistic and antagonistic interactions with the host immune system. Front Microbiol. 2018; 8: 2521.
[60] Engdahl E, Gustafsson R, Huang J, et al. Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. Front Immunol. 2019; 10: 2715.
[61] Hogestyn JM, Mock DJ, Mayer-Proschel M. Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology. Neural Regen Res. 2018; 13 (2): 211.
[62] Xie J, Tian S, Liu J, et al. Dual role of the nasal microbiota in neurological diseases. An unignorable risk factor or a potential therapy carrier. Pharmacol Res. 2022; 106189.
[63] Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol. 2003; 53 (2): 189-197.
[64] Hogestyn JM, Mock DJ, Mayer-Proschel M. Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology. Neural Regen Res. 2018; 13 (2): 211.
[65] Hayman IR, Temple RM, Burgess CK, et al. New insight into Epstein-Barr Virus infection using models of stratified epithelium. PLoS Pathog. 2023; 19 (1): e1011040.
[66] Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022; 375 (6578): 296-301.
[67] Houen G, Trier NH, Frederiksen JL. Epstein-Barr virus and multiple sclerosis. Front Immunol. 2020; 11: 587078.
[68] Meier U-C, Cipian RC, Karimi A, Ramasamy R, Middeldorp JM. Cumulative roles for Epstein-Barr virus, human endogenous retroviruses, and human herpes virus-6 in driving an inflammatory cascade underlying MS pathogenesis. Front Immunol. 2021; 4526.
[69] Veroni C, Aloisi F. The CD8 T cell-Epstein-Barr virus-B cell trialogue: a central issue in multiple sclerosis pathogenesis. Front Immunol. 2021; 12: 665718.
[70] Nielsen T, Rostgaard K, Askling J, et al. Effects of infectious mononucleosis and HLA-DRB1* 15 in multiple sclerosis. Mult Scler. 2009; 15 (4): 431-436.
[71] Keane JT, Afrasiabi A, Schibeci SD, Swaminathan S, Parnell GP, Booth DR. The interaction of Epstein-Barr virus encoded transcription factor EBNA2 with multiple sclerosis risk loci is dependent on the risk genotype. EBioMedicine. 2021; 71: 103572.
[72] Bar-Or A, Pender MP, Khanna R, et al. Epstein–Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol Med. 2020; 26 (3): 296-310.
[73] Van Langelaar J, Rijvers L, Smolders J, Van Luijn MM. B and T cells driving multiple sclerosis: identity, mechanisms and potential triggers. Front Immunol. 2020; 11: 760.
[74] Zdimerova H, Murer A, Engelmann C, et al. Attenuated immune control of Epstein–Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol. 2021; 51 (1): 64-75.
[75] Lazibat I, Rubinić-Majdak M, Županić S. Multiple sclerosis: new aspects of immunopathogenesis. Acta Clin Croat. 2018; 57 (2): 352.
[76] Lee DS, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov. 2021; 20 (3): 179-199.
[77] Zivadinov R, Ramanathan M, Hagemeier J, et al. Teriflunomide’s effect on humoral response to Epstein-Barr virus and development of cortical gray matter pathology in multiple sclerosis. Mult Scler Relat Disord. 2019; 36: 101388.
[78] Bjornevik K, Münz C, Cohen JI, Ascherio A. Epstein–Barr virus as a leading cause of multiple sclerosis: mechanisms and implications. Nat Rev Neurol. 2023; 19 (3): 160-171.
[79] Perron H, Garson J, Bedin F, et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. Proc Natl Acad Sci USA. 1997; 94 (14): 7583-7588.
[80] Laufer G, Mayer J, Mueller BF, Mueller-Lantzsch N, Ruprecht K. Analysis of transcribed human endogenous retrovirus W env loci clarifies the origin of multiple sclerosis-associated retrovirus envsequences. Retrovirology. 2009; 6: 1-17.
[81] Chen J, Foroozesh M, Qin Z. Transactivation of human endogenous retroviruses by tumor viruses and their functions in virus-associated malignancies. Oncogenesis. 2019; 8 (1): 6.
[82] Ahangar Oskouee M, Shahmahmoodi S, Jalilvand S, et al. No evidence of mammary tumor virus env gene-like sequences among Iranian women with breast cancer. Intervirology. 2014; 57 (6): 353-356.
[83] Ko E-J, Song KS, Ock MS, et al. Expression profiles of human endogenous retrovirus (HERV)-K and HERV-R Env proteins in various cancers. BMB Rep. 2021; 54 (7): 368.
[84] Steiner MC, Marston JL, Iñiguez LP, et al. Locus-Specific characterization of human endogenous retrovirus expression in prostate, breast, and colon cancers HERV expression in prostate, breast, and colon cancers. Cancer Res. 2021; 81 (13): 3449-3460.
[85] Kristensen MK, Christensen T. Regulation of the expression of human endogenous retroviruses: elements in fetal development and a possible role in the development of cancer and neurological diseases. APMIS. 2021; 129 (5): 241-253.
[86] Zhang M, Liang JQ, Zheng S. Expressional activation and functional roles of human endogenous retroviruses in cancers. Rev Med Virol. 2019; 29 (2): e2025.
[87] Morris G, Maes M, Murdjeva M, Puri BK. Do human endogenous retroviruses contribute to multiple sclerosis, and if so, how? Mol Neurobiol. 2019; 56 (4): 2590-2605.
[88] Arru G, Leoni S, Pugliatti M, et al. Natalizumab inhibits the expression of human endogenous retroviruses of the W family in multiple sclerosis patients: a longitudinal cohort study. Mult Scler. 2014; 20 (2): 174-182.
[89] Arru G, Sechi E, Mariotto S, et al. Antibody response against HERV-W env surface peptides differentiates multiple sclerosis and neuromyelitis optica spectrum disorder. Mult Scler J Exp Transl Clin. 2017; 3 (4): 2055217317742425.
[90] Mameli G, Madeddu G, Mei A, et al. Activation of MSRV-type endogenous retroviruses during infectious mononucleosis and Epstein-Barr virus latency: the missing link with multiple sclerosis? PLoS One, 2013; 8 (11): e78474.
[91] Kremer D, Gruchot J, Weyers V, et al. pHERV-W envelope protein fuels microglial cell-dependent damage of myelinated axons in multiple sclerosis. Proc Natl Acad Sci USA. 2019; 116 (30): 15216-15225.
[92] Morandi E, Tarlinton RE, Tanasescu R, Gran B. Human endogenous retroviruses and multiple sclerosis: causation, association, or after-effect? Mult Scler. 2017; 23 (8): 1050-1055.
[93] Giménez-Orenga K, Oltra E. Human endogenous retrovirus as therapeutic targets in neurologic disease. Pharmaceuticals (Basel). 2021; 14 (6): 495.
[94] Bu C, Wang Z, Ren Y, Chen D, Jiang S-W. Sciences, M.L. Syncytin-1 nonfusogenic activities modulate inflammation and contribute to preeclampsia pathogenesis. Cell Mol Life Sci. 2022; 79 (6): 290.
[95] Sheng L, Luo Q, Chen L. Disposition. Amino acid solute carrier transporters in inflammation and autoimmunity. Drug Metab Dispos. 2022; 50: 1228-1237.
[96] Mechelli R, Carmela R, Roberta R, et al. Neuroinflammation. Viruses and neuroinflammation in multiple sclerosis. Neuroimmunol Neuroinflamm. 2021.
[97] Mosser JF, Grant LR, Millar EV, et al. Nasopharyngeal carriage and transmission of Streptococcus pneumoniae in American Indian households after a decade of pneumococcal conjugate vaccine use. PLoS One. 2014; 9 (1): e79578.
[98] Tarlinton RE, Martynova E, Rizvanov AA, Khaiboullina S, Verma S. Role of viruses in the pathogenesis of multiple sclerosis. Viruses. 2020; 12 (6): 643.
[99] Annibali V, Mechelli R, Romano S, et al. IFN-β and multiple sclerosis: from etiology to therapy and back. Cytokine Growth Factor Rev. 2015; 26 (2): 221-228.
[100] Cohen M, De Sèze J, Marignier R, Lebrun C. False positivity of anti aquaporin-4 antibodies in natalizumab-treated patients. Mult Scler J. 2016; 22 (9): 1231-1234.
[101] Chen Y-H, Lightman S, Eskandarpour M, Calder VL. Adhesion molecule targeted therapy for non-infectious uveitis. Int J Mol Sci. 2022; 23 (1): 503.
[102] Dsilva L, McCarthy K, Lyons J, et al. Progressive multifocal leukoencephalopathy with natalizumab extended or standard interval dosing in the United States and the rest of the world. Expert Opin Drug Saf. 2023; 22 (10): 995-1002.